Charcot-Marie-Tooth disease (CMT) adalah salah satu penyakit saraf tepi (peripheral neuropathy) bawaan yang paling umum dijumpai, namun ironisnya masih sering terlambat terdiagnosis. Kondisi ini dinamai berdasarkan tiga dokter yang pertama kali mendeskripsikannya secara independen pada tahun 1886: Jean-Martin Charcot dan Pierre Marie dari Prancis, serta Howard Henry Tooth dari Inggris. Meski bukan nama yang mudah diucapkan dalam percakapan sehari-hari, penyakit ini relevan untuk diketahui oleh tenaga kesehatan maupun masyarakat umum, karena dampaknya pada kualitas hidup penyintas bersifat signifikan dan berlangsung seumur hidup.
Seberapa Umum CMT?
CMT bukan penyakit langka dalam arti sesungguhnya. Berdasarkan tinjauan komprehensif yang diterbitkan di Nature Reviews Disease Primers tahun 2026, CMT dan neuropati terkait diperkirakan menyerang sekitar 1 dari 2.500 orang di seluruh dunia, mengenai pria dan wanita tanpa perbedaan bermakna (Burns et al., 2026). Angka ini menjadikan CMT sebagai salah satu penyakit neuromuskular herediter paling predominan secara global, termasuk dalam kelompok kelainan neurologis yang diturunkan.
Data dari Registri CMT Italia yang melibatkan lebih dari 1.000 pasien menunjukkan bahwa bentuk demielinasi (CMT1) merupakan yang paling sering dijumpai (65,3%), diikuti bentuk aksonal (CMT2, 24,6%), dan bentuk intermediat (9,0%). Mutasi duplikasi gen PMP22 adalah penyebab tersering, mencakup sekitar 45% kasus dengan diagnosis genetik yang terkonfirmasi (Pisciotta et al., 2023). Di Asia, termasuk Indonesia, data epidemiologi yang spesifik masih terbatas, namun pola genetik yang ditemukan di populasi Asia Timur memberikan gambaran bahwa penyakit ini juga ada di antara kita, hanya saja belum terdata dengan baik.
Apa yang Terjadi pada Saraf Penderita CMT?
Untuk memahami CMT, perlu dipahami terlebih dahulu anatomi dasar saraf tepi. Sel saraf (neuron) memiliki akson, yaitu serabut panjang yang menghantarkan sinyal listrik. Pada banyak saraf, akson ini dibungkus oleh selubung mielin—semacam pelapis isolasi yang dibentuk oleh sel Schwann—yang memungkinkan penghantaran impuls saraf berlangsung cepat dan efisien.
CMT mengganggu sistem ini melalui dua mekanisme utama yang berbeda tergantung subtipenya. Pertama, pada bentuk demielinasi (CMT1), mutasi gen menyebabkan kerusakan atau disfungsi selubung mielin itu sendiri, sehingga penghantaran sinyal melambat drastis. Kedua, pada bentuk aksonal (CMT2), kerusakan terjadi langsung pada akson saraf tanpa mengenai mielin secara primer, dengan mekanisme yang seringkali melibatkan disfungsi mitokondria, gangguan pada sitoskeleton aksonal, atau mutasi pada enzim aminoasil-tRNA sintetase (Burns et al., 2026).
Keduanya berujung pada satu proses yang sama: degenerasi aksonal yang progresif dan bergantung pada panjang saraf (length-dependent axonal loss). Artinya, saraf yang paling panjang—yaitu yang menjangkau ujung kaki dan tangan—mengalami kerusakan paling awal dan paling berat. Inilah yang menjelaskan pola gejala khas CMT yang muncul dari distal ke proksimal.
Mengenali Wajah Klinis CMT
Gejala CMT bervariasi cukup lebar antarpasien, bahkan di dalam keluarga yang sama, namun terdapat pola klinis yang relatif konsisten.
Kelemahan otot distal adalah keluhan yang paling sering membawa pasien ke dokter. Penderita biasanya mengeluhkan kesulitan berjalan, sering tersandung, atau tidak mampu mengangkat kaki bagian depan (foot drop). Kelemahan ini secara khas dimulai dari otot-otot kaki bagian bawah, terutama otot yang bertugas mendorsifleksikan pergelangan kaki. Seiring waktu, tangan dan jari-jari pun ikut melemah, menyulitkan aktivitas seperti membuka botol, mengancingkan baju, atau menggenggam benda kecil.
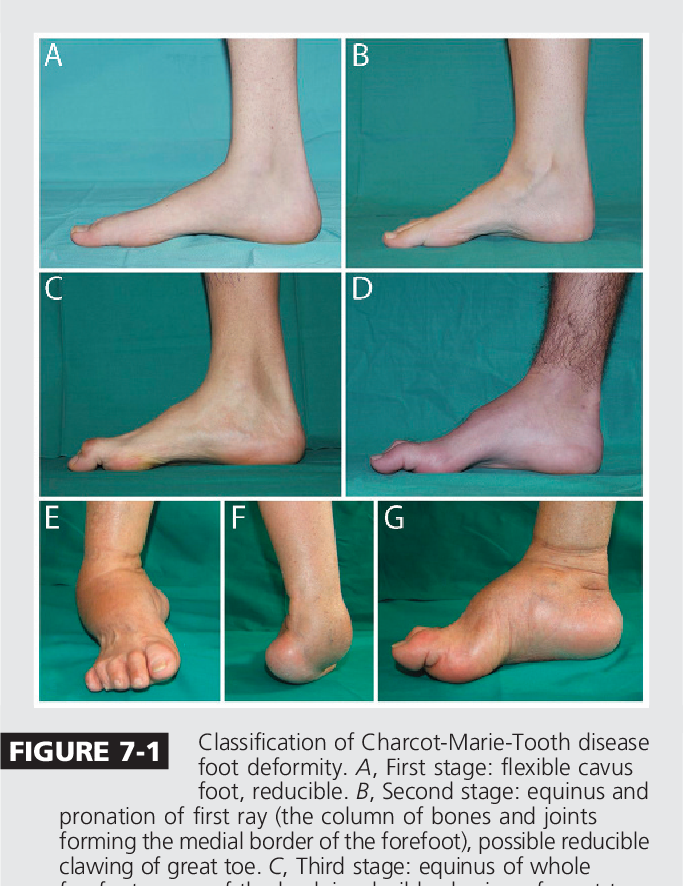
Deformitas muskuloskeletal merupakan konsekuensi kelemahan kronik yang tidak seimbang. Kaki cekung yang berlebihan (pes cavus) dan jari-jari kaki seperti palu (hammer toes) adalah temuan klasik yang dapat terlihat bahkan pada anak-anak. Skoliosis (kelengkungan tulang belakang) ditemukan pada sekitar 20% pasien dalam registri Italia, terutama pada bentuk CMT resesif (Pisciotta et al., 2023). Pada CMT onset dini, displasia panggul juga dapat terjadi.
Refleks tendon yang menghilang secara menyeluruh, terutama refleks Achilles, merupakan salah satu tanda klinis paling awal yang dapat dideteksi oleh dokter bahkan sebelum gejala fungsional muncul secara nyata.
Gangguan sensoris distal berupa rasa kebas, kesemutan, atau berkurangnya persepsi terhadap sentuhan ringan, nyeri, dan suhu di ujung kaki dan tangan turut mewarnai gambaran klinis CMT, meski derajatnya bervariasi antarpasien.
Secara keseluruhan, dibandingkan populasi umum, anak-anak maupun dewasa dengan CMT memiliki kualitas hidup yang lebih rendah di domain fisik, emosional, maupun sosial—dengan domain fisik sebagai yang paling memberatkan (Burns et al., 2026).
Dasar Genetik: Ribuan Cara Untuk Sakit Satu Penyakit
Salah satu aspek yang membuat CMT sangat menantang secara ilmiah adalah heterogenitas genetiknya yang luar biasa. Diperkirakan lebih dari 100 lokus pada genom manusia telah dikaitkan dengan berbagai bentuk CMT dan neuropati herediter terkait (Okamoto & Takashima, 2023). Masing-masing gen yang bermutasi memberikan dampak pada protein yang berbeda, sehingga mekanisme kerusakan pun bervariasi.
Sebagai contoh, gen PMP22 yang mengalami duplikasi pada kromosom 17 menyebabkan kelebihan produksi protein mielin perifer 22, yang kemudian mengganggu pembentukan mielin—ini adalah penyebab CMT1A, subtipe paling umum. Sementara itu, mutasi pada gen GJB1 (yang mengkode protein koneksin-32) menyebabkan CMT terkait kromosom X (CMTX1), suatu bentuk yang memiliki pola pewarisan khas: pria umumnya lebih terdampak dibandingkan perempuan yang hanya menjadi pembawa (carrier).
Beberapa gen lain yang dikenal luas dalam konteks CMT adalah MPZ (protein myelin P0), MFN2 (mitofusin-2, terkait fungsi mitokondria pada CMT2A), HSPB1 (protein kejut panas kecil), dan NEFL (rantai ringan neurofilamen). Setiap mutasi memiliki “sidik jari” klinisnya masing-masing, meskipun terdapat tumpang-tindih yang cukup besar.
Penemuan terbaru yang menarik perhatian komunitas neurologi global adalah CMT-SORD, bentuk CMT aksonal resesif yang disebabkan oleh mutasi loss-of-function bialelik pada gen SORD (sorbitol dehidrogenase). Sebuah studi multisenter besar yang melibatkan 144 pasien dari 126 keluarga di berbagai negara menunjukkan bahwa CMT-SORD merupakan salah satu bentuk CMT resesif yang paling umum, dengan onset gejala biasanya pada dekade kedua kehidupan dan dominasi kelemahan motorik (Cortese et al., 2025). Pentingnya CMT-SORD semakin menonjol karena bentuk ini memiliki biomarker yang terukur (kadar sorbitol serum) dan menjadi target terapi obat yang sedang dalam uji klinis.
Bagaimana CMT Didiagnosis?
Diagnosis CMT idealnya merupakan kombinasi dari evaluasi klinis yang cermat, pemeriksaan neurofisiologi, dan konfirmasi genetik.
Evaluasi klinis dimulai dari anamnesis keluarga yang mendetail. CMT umumnya memiliki pola pewarisan dominan autosomal, resesif autosomal, atau terkait kromosom X, sehingga riwayat anggota keluarga dengan keluhan serupa—meski sering dianggap “hanya kebiasaan keluarga yang bertelanjang kaki”—memberikan petunjuk penting.
Studi konduksi saraf (nerve conduction study, NCS) dan elektromielografi (EMG) sangat membantu dalam mengklasifikasikan CMT. Pada CMT1 (demielinasi), kecepatan konduksi motorik melambat secara bermakna (biasanya di bawah 38 m/detik pada saraf median). Pada CMT2 (aksonal), kecepatan konduksi relatif normal namun amplitudo potensial aksi menurun, mencerminkan berkurangnya jumlah akson yang berfungsi.
Pengujian genetik merupakan baku emas untuk konfirmasi diagnosis dan penentuan subtipe. Teknologi next-generation sequencing (NGS), termasuk panel gen herediter neuropati dan whole-exome sequencing, telah merevolusi kemampuan diagnostik CMT dalam satu dekade terakhir. Namun demikian, akses terhadap pemeriksaan genetik ini masih terbatas di banyak negara berkembang, termasuk Indonesia, mengingat biayanya yang tidak murah dan belum semua fasilitas kesehatan memiliki kapasitas tersebut.
Penatalaksanaan: Fokus pada Fungsi, Bukan Hanya Obat
Hingga saat ini, tidak ada terapi yang mampu memodifikasi perjalanan penyakit CMT pada tingkat yang sudah disetujui secara klinis. Penatalaksanaan saat ini berfokus pada rehabilitasi, ortotik, intervensi bedah selektif, dan penanganan gejala yang menyertai (Beloribi-Djefaflia & Attarian, 2022; De Grado et al., 2025).
Rehabilitasi fisik merupakan pilar utama manajemen CMT. Physiotherapy dan terapi okupasi bertujuan mempertahankan kekuatan otot, meningkatkan keseimbangan dan mobilitas, serta mencegah kontraktur sendi. Tinjauan sistematis uji klinis acak menunjukkan bahwa latihan penguatan dorsiflexor pergelangan kaki memberikan bukti moderat dalam memperlambat progresi kelemahan selama 24 bulan pada anak-anak dengan CMT1A. Namun secara umum, kualitas bukti untuk berbagai modalitas latihan pada CMT masih berada di level rendah hingga sedang, sehingga diperlukan uji klinis yang lebih robust (Conde et al., 2023).
Ortotik memiliki peran yang sangat praktis dalam keseharian penyintas CMT. Ankle foot orthosis (AFO) membantu mengompensasi kelemahan otot penyangga kaki, sehingga pasien dapat berjalan dengan lebih aman dan mengurangi risiko jatuh. Data registri Italia menunjukkan bahwa hampir separuh pasien CMT menggunakan alat bantu ortotik, dengan mayoritas mulai memakainya pada usia 30-an (Pisciotta et al., 2023).
Panduan klinis praktik pediatrik yang diterbitkan pada 2022 oleh konsorsium internasional mencakup 34 rekomendasi (3 berbasis bukti, 31 berbasis konsensus) yang mencakup penanganan kelemahan otot, gangguan keseimbangan, gejala sensoris, kram otot, gangguan fungsi ekstremitas atas, gangguan respirasi, serta pemeliharaan lingkup gerak sendi pada anak-anak (Yiu et al., 2022).
Intervensi bedah pada deformitas kaki, seperti koreksi pes cavus berat atau transfer tendon, dapat dipertimbangkan pada kasus tertentu untuk meningkatkan fungsi berjalan, meski keputusan ini memerlukan evaluasi multidisiplin yang hati-hati.
Cakrawala Terapi Baru: Antara Harapan dan Tantangan
Lanskap terapi CMT sedang berubah. Kemajuan dalam memahami mekanisme penyakit di tingkat molekuler telah membuka berbagai jalur terapeutik yang menjanjikan, meski sebagian besar masih dalam tahap praklinis atau uji klinis awal (Okamoto & Takashima, 2023; Dong et al., 2024).
PXT3003 adalah kombinasi dari tiga obat dosis rendah (baklofen, naltrexon, dan sorbitol) yang telah menjalani uji klinis fase 3 untuk CMT1A. Obat ini bekerja dengan menekan ekspresi berlebih protein PMP22, dan menjadi salah satu kandidat terapi farmakologis yang paling maju saat ini.
Govorestat (inhibitor aldosa reduktase) diposisikan sebagai terapi potensial pertama yang secara spesifik menarget CMT-SORD. Analisis sementara dari uji klinis fase 2/3 sedang dinantikan dengan penuh antusiasme oleh komunitas ilmiah, dengan potensi menjadi obat CMT pertama yang mendapatkan persetujuan regulasi (De Grado et al., 2025).
Terapi gen menawarkan paradigma yang berbeda: alih-alih memodifikasi perjalanan penyakit secara tidak langsung, terapi gen bertujuan mengoreksi akar masalah di tingkat DNA. Strategi yang sedang dikembangkan meliputi gene silencing menggunakan antisense oligonucleotides (ASOasi) untuk menekan ekspresi PMP22 berlebih pada CMT1A, gene addition untuk mengekspresikan GJB1 yang fungsional pada CMTX1, serta gene editing berbasis CRISPR. Saat ini, terapi gen untuk subtipe CMT2S adalah yang paling maju secara klinis (Dong et al., 2024). Meski demikian, tantangan berupa keamanan, efisiensi penghantaran ke sel saraf tepi, dan skalabilitas produksi masih perlu diatasi sebelum terapi ini menjadi kenyataan klinis yang luas.
Inhibitor HDAC6 adalah kelas obat baru yang bekerja dengan memperbaiki transportasi aksonal yang terganggu, suatu mekanisme patologis yang ditemukan pada berbagai subtipe CMT aksonal. Obat-obat ini sedang mendekati fase uji klinis (De Grado et al., 2025).
Satu pendekatan yang lebih mendasar namun menarik adalah penelitian mengenai peran transporter kolesterol ABCA1 (ATP-binding cassette A1) dalam memperbaiki biosintesis mielin di saraf tepi. Studi praklinis pada model hewan CMT tipe 1 menunjukkan ABCA1 sebagai target terapeutik potensial dalam upaya perbaikan mielin (Woo et al., 2026), meskipun penerapan klinisnya masih jauh.
Perspektif Pasien dan Layanan di Indonesia
CMT adalah penyakit yang berlangsung seumur hidup. Penyintas CMT harus belajar beradaptasi dengan perubahan kemampuan fisik yang progresif, dan ini berdampak tidak hanya pada mobilitas, tetapi juga pada pekerjaan, hubungan sosial, dan kesehatan mental. Dukungan psikososial, konseling genetik bagi keluarga yang merencanakan kehamilan, dan akses terhadap perawatan multidisiplin merupakan kebutuhan yang sama pentingnya dengan penanganan fisik.
Di Indonesia, CMT masih termasuk dalam kategori penyakit langka yang pengelolaannya belum memiliki panduan atau jalur rujukan yang baku. Akses ke spesialis neurologi dan fasilitas pemeriksaan neurofisiologi sudah semakin membaik di kota-kota besar, meski belum merata. Pengujian genetik yang terjangkau masih menjadi tantangan utama. Dalam kerangka JKN (Jaminan Kesehatan Nasional), terapi rehabilitasi fisik dapat diakses melalui poli fisioterapi di fasilitas kesehatan tingkat lanjut, meski koordinasi antardisiplin masih perlu diperkuat.
Registri pasien CMT yang komprehensif—serupa dengan yang telah berhasil dibangun di Italia atau dalam jaringan internasional—kiranya menjadi langkah strategis yang perlu dipikirkan oleh komunitas neurologi Indonesia untuk memahami beban sesungguhnya dari penyakit ini di populasi kita.
Kesimpulan
Charcot-Marie-Tooth disease adalah neuropati herediter yang paling umum, namun terlalu sering terlambat dikenali. Heterogenitas genetiknya yang ekstrem menjadikannya tantangan diagnostik sekaligus medan riset yang aktif dan dinamis. Meski hingga kini belum ada terapi yang menyembuhkan, pendekatan rehabilitatif yang terstruktur dan penggunaan ortotik yang tepat mampu secara bermakna mempertahankan fungsi dan kualitas hidup pasien. Di cakrawala, terapi gen dan obat-obat yang menarget mekanisme spesifik CMT membuka harapan nyata bahwa dalam satu dekade ke depan, pasien CMT akhirnya akan memiliki pilihan terapi yang memodifikasi perjalanan penyakit—bukan sekadar mengelola gejalanya.
Daftar Referensi
Beloribi-Djefaflia, S., & Attarian, S. (2022). Treatment of Charcot-Marie-Tooth neuropathies. Revue Neurologique, 179(1–2), 35–48. https://doi.org/10.1016/j.neurol.2022.11.006
Burns, J., Timmerman, V., Laurá, M., Yiu, E. M., D’Antonio, M., Mukherjee-Clavin, B., De Winter, J., & Scherer, S. S. (2026). Charcot-Marie-Tooth disease and related neuropathies. Nature Reviews Disease Primers, 12(1), 3. https://doi.org/10.1038/s41572-025-00679-2
Conde, R. M., Senem, I., dos Santos, M., de Lima Osório, F., & Marques Júnior, W. (2023). Effectiveness of exercise therapy for individuals diagnosed with Charcot-Marie-Tooth disease: A systematic review of randomized clinical trials. Journal of the Peripheral Nervous System, 28(2), 169–178. https://doi.org/10.1111/jns.12548
Cortese, A., Dohrn, M. F., Curro, R., Negri, S., Lassuthova, P., Pisciotta, C., … Shy, M. E. (2025). Genotype and phenotype spectrum of Charcot-Marie-Tooth disease due to mutations in SORD. Brain, 148(10), 3737–3747. https://doi.org/10.1093/brain/awaf021
De Grado, A., Serio, M., Saveri, P., Pisciotta, C., & Pareyson, D. (2025). Charcot-Marie-Tooth disease: A review of clinical developments and its management — What’s new in 2025? Expert Review of Neurotherapeutics, 25(4), 427–442. https://doi.org/10.1080/14737175.2025.2470980
Dong, H., Qin, B., Zhang, H., Lei, L., & Wu, S. (2024). Current treatment methods for Charcot-Marie-Tooth diseases. Biomolecules, 14(9), 1138. https://doi.org/10.3390/biom14091138
Okamoto, Y., & Takashima, H. (2023). The current state of Charcot-Marie-Tooth disease treatment. Genes, 14(7), 1391. https://doi.org/10.3390/genes14071391
Pan, X., Xie, J., Li, Z., Xiang, Y., Yu, Y., Cai, Q., Xu, H., Wan, Y., & Xing, J. (2026). Stress-driven selective neuronal vulnerability in Charcot-Marie-Tooth disease: From prodromal pathology to therapeutic implications. Cells, 15(3), 271. https://doi.org/10.3390/cells15030271
Pisciotta, C., Bertini, A., Tramacere, I., Manganelli, F., Fabrizi, G. M., Schenone, A., … Pareyson, D. (2023). Clinical spectrum and frequency of Charcot-Marie-Tooth disease in Italy: Data from the National CMT Registry. European Journal of Neurology, 30(8), 2461–2470. https://doi.org/10.1111/ene.15860
Woo, Y. H., Schmidt, N. E., Johansson, J. O., & Notterpek, L. (2026). ABCA1: A therapeutic target for improving cholesterol homeostasis in peripheral neuropathies. Biomolecules, 16(2), 332. https://doi.org/10.3390/biom16020332
Yiu, E. M., Bray, P., Baets, J., Baker, S. K., Barisic, N., de Valle, K., … Burns, J. (2022). Clinical practice guideline for the management of paediatric Charcot-Marie-Tooth disease. Journal of Neurology, Neurosurgery & Psychiatry, 93(5), 530–538. https://doi.org/10.1136/jnnp-2021-328483
Artikel ini disusun berdasarkan tinjauan literatur ilmiah terkini dari PubMed dan sumber akademik terpercaya. Informasi dalam artikel ini bersifat edukatif dan tidak menggantikan konsultasi dengan tenaga medis profesional.

Tinggalkan komentar